

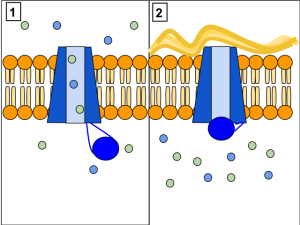
Das CFTR (Cystic Fibrosis Transmembrane Conductance Regulator)-Protein reguliert den Wasser- und Salztransport in der Plasmamembran und in Epithelzellen (1).
CFTR-Mutationen (2), wie sie bei Zystischer Fibrose auftreten, behindern den Chloridionen-Transport aus der Zelle oder blockieren ihn sogar völlig. Dadurch entsteht ein Konzentrationsunterschied, bei dem viele Ionen in der Zelle vorliegen, aber nur wenige im Sekret vorhanden sind. Wegen der dadurch in der Zelle auftretenden osmotischen Kräfte wird dem Sekret Wasser entzogen. Das Sekret wird zähflüssig, kann dadurch auch nur schlecht abgebaut werden und verstopft feine Kanäle, wie sie etwa in der Lunge vorkommen. © Lbudd14. CC BY-SA 3.0.
Die Cystische Fibrose ist eine häufige, angeborene Erkrankung der Lunge und des Verdauungssystems der ein Gendefekt zugrunde liegt. Wissenschaftler konnten nun zeigen, dass bei einem großen Teil der erwachsenen Patienten mit Cystischer Fibrose eine zusätzlich Störung des Immunsystems vorliegt: Ein für die Immunabwehr wichtiges Oberflächenmolekül ist bei ihnen vermindert oder fehlt ganz.
Bei der Cystischen Fibrose (CF, auch bekannt als Mukoviszidose) führt die Mutation eines Ionenkanals zu einer veränderten Zusammensetzung von Körpersekreten. In der Folge bildet sich ein zähflüssiger Schleim, der Störungen in Lunge und Magen-Darm-Trakt hervorruft. Außerdem leiden die Patienten sehr häufig an hartnäckigen Infektionen. Das Team um Loems Ziegler-Heitbrock vom Helmholtz Zentrum München untersuchte daher, ob bei den Patienten eine zusätzliche Immunstörung vorliegt. Die Wissenschaftler konnten zeigen, dass ein für die Immunantwort wichtiges Oberflächen-Molekül, HLA-DQ, bei einem großen Teil der Patienten in verminderter Menge vorhanden ist oder fehlt.
Alle wichtigen Immunzellen sind von der Veränderung betroffen
Die HLA-DQ Moleküle sind Oberflächen-Proteine (MHC-Klasse II), die einzelne Bestandteile von eindringenden Mikroorganismen präsentieren, um damit Immunzellen zu aktivieren, die diese Erreger dann gezielt eliminieren. Diese Oberflächen-Proteine finden sich insbesondere auf primären Immunzellen, wie den Monozyten, den Makrophagen und den so genannten dendritischen Zellen. Die Wissenschaftler konnten zeigen, dass die HLA-DQ Proteine auf allen diesen Zellen sowohl im Blut als auch in der Lunge vermindert sind oder fehlen können. Damit gilt diese Störung für alle wichtigen präsentierenden Zellen des Immunsystems.
Genauer molekularer Mechanismus soll noch geklärt werden
Um zu verstehen, wie diese Störung zustande kommt, haben die Wissenschaftler die einzelnen Schritte der molekularen Regulation untersucht. Sie konnten zeigen, dass bei den betroffenen CF-Patienten der Entzündungs-Botenstoff Interferon-gamma nicht in der Lage ist, das Signalüberträger-Molekül CIITA zu induzieren. In der Folge unterbleibt die Bildung von HLA-DQ. Zu klären bleibt, warum Interferon-gamma das CIITA Molekül nicht aktiviert und in welchem Maße diese Störung zum Verlauf der Erkrankung beiträgt. In weiteren Untersuchungen wollen die Wissenschaftler einen schnellen Test entwickeln, um die Immunstörung nachzuweisen. Die Dysfunktion der körpereigenen Abwehr könnte für die Diagnostik und Behandlung von Cystischer Fibrose von großer Bedeutung sein.
Helmholtz Zentrum München, 04.09.2014.
Original-Publikation:
Hofer, TPJ et al (2014). Decreased Expression of HLA-DQ and -DR on cells of the monocytic lineage in cystic fibrosis, Journal of Molecular Medicine, doi: 10.1007/s00109-014-1200-z