

Aufbau einer Nervenzelle. © Quasar Jarosz. CC BY-SA 3.0.
Mutierte Prion-Proteine lösen bei Rindern BSE und beim menschen die Creutzfeldt-Jakob-Krankheit aus. Obwohl diese Proteine in fast jedem Organismus vorkommen, glaubte man lange, dass die Proteine keine wichtige Funktion erfüllen und ein möglicher Therapieansatz darin bestünde, sie zu eliminieren. Doch, wie Forscher nun entdeckt haben sind diese Eiweißmoleküle keineswegs entbehrlich, sondern sorgen gemeinsam mit einem Rezeptor für die Gesundheit der Nerven. Aus diesen Erkenntnissen könnten sich neue Therapien für chronische Nervenkrankheiten ergeben.
Seit der Entdeckung des Prion-Gens im Jahr 1985 rätseln Forscher über dessen natürliche Rolle in Nervenzellen. «Wir können dem Prion-Protein nun endlich eine klar umrissene Funktion zuweisen und aufzeigen, dass es im Verbund mit einem bestimmten Rezeptor für die langfristige Intaktheit der Nerven zuständig ist», sagt Adriano Aguzzi vom Neuropathologischen Institut der Universität Zürich.
Mutierte Prionen können bei Mensch und Tier fatale Degenerationen des Gehirns auslösen. In den 1990er Jahren sorgten sie als die Auslöser des Rinderwahnsinns in den Medien für Schlagzeilen. Beim Menschen führen sie zur Creutzfeldt-Jakob-Krankheit und anderen neurologischen Störungen, die bis heute nicht therapierbar sind und nach wie vor tödlich verlaufen. Die infektiösen Prionen bestehen aus einer falsch gefalteten Form eines normalen Prion-Proteins PrPC, das in der Membran der Nervenzellen vorkommt. Die infektiösen Prionen vermehren sich, indem sie das normale Protein in weitere infektiöse Prionen umwandeln.
Ohne Prion-Proteine entstehen Nervenkrankheiten
Lange rätselten die Forscher, warum unsere Nervenzellen ein Protein bilden, das scheinbar keine wichtige Funktion besitzt, aber bei falscher Faltung höchst gefährlich werden kann. Als Prionenforscher beschäftigt Aguzzi diese Frage seit Jahrzehnten und er stellte die Hypothese auf, dass Tiere, die das PrPC-Gen nicht besitzen, resistent gegen Prionen-Erkrankungen sein könnten. Doch wie wirkt sich das Fehlen des Prion-Proteins auf den Organismus aus?
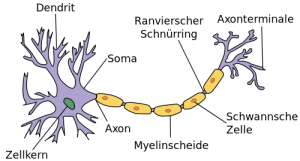
Bereits vor einige Jahren fanden Aguzzi mit seinem Team heraus, dass Mäuse ohne PrPC-Gen an einer chronischen Erkrankung der peripheren Nerven leiden. Denn die sogenannten Schwann-Zellen die die empfindlichen Nervenfasern umschließen bilden keine elektrische Isolationsschicht mehr, um diese zu schützen. Durch dieses Defizit an isolierendem Myelin erkranken die peripheren Nerven und es kann zu motorischen Störungen des Bewegungsapparates und zu Lähmungen kommen.
Als nächstes untersuchte das Forscherteam, warum die peripheren Nerven geschädigt werden, wenn kein Prion-Protein PrPC vorhanden ist. Dabei entdeckten sie, wie das von den Nervenzellen gebildete PrPC an die Schwann-Zellen bindet: nämlich über einen Rezeptor namens «Gpr126». Durch die Bindung des Prion-Proteina an den Rezeptor wird der Botenstoff cAMP freigesetzt, der für das Aufrechterhalten der Nervenschutzhülle essentiell ist. «Gpr126» gehört zur umfangreichen Familie der «G-Protein-gekoppelten» Rezeptoren, die an vielen physiologischen Prozessen beteiligt sind. Fehlfunktionen dieser Rezeptoren sind Ursache zahlreicher Erkrankungen.
30 Jahre alte Forschungsfrage endlich geklärt
Die neuen Erkenntnisse klären eine wichtige Frage, die Neurowissenschaftler schon lange umtreibt. «Will man bei möglichen Therapien gegen die Creutzfeldt-Jakob-Krankheit das Prion-Protein PrPC ganz ausschalten, muss man sich künftig der potentiellen Nebenwirkung auf die Nerven bewusst sein», erklärt Aguzzi. Zudem könnte sich aus den vorliegenden Erkenntnissen über die Wirkungsweise des PrPC auf molekularer Ebene ein neuer Therapieansatz für periphere Neuropathien ergeben. Für diese belastenden chronische Erkrankungen des Nervensystems gibt es derzeit nur sehr eingeschränkte Therapiemöglichkeiten.
Universität Zürich, 6. August 2016
Originalpublikation:
Alexander Küffer, Asvin K. K. Lakkaraju, Amit Mogha, Sarah C. Petersen, Kristina Airich, Cédric Doucerain, Rajlakshmi Marpakwar, Pamela Bakirci, Assunta Senatore, Arnaud Monnard, Carmen Schiavi, Mario Nuvolone, Bianka Grosshans, Simone Hornemann, Frederic Bassilana, Kelly R. Monk & Adriano Aguzzi. The prion protein is an agonistic ligand of the G-protein-coupled receptor Gpr1/Adgrg6. Nature, 8 August 2016. doi: 10.1038/nature19312